




Google Scholar citation report
Citations : 25586
Systematic Reviews in Pharmacy received 25586 citations as per google scholar report

Systematic Reviews in Pharmacy peer review process verified at publons

Indexed In
Research Article - (2022) Volume 13, Issue 1
Abstract
Electron microscopy tomography results obtained after increasing the resistance of flash-frozen proteins to accelerated electron beams. This curing reduces electron dose dependence, allowing high-resolution tomography without need of averaging multiple samples. Three-dimensional images at atomic-scale are realized on one individual protein in one experiment. This workflow, introduced for flash-frozen rigid biopolymers, is here generalized and applied to flash-frozen, deformable oligomers extracted from a virus. Results are shown down to atomic coordinates without recurring to averaging among many samples. It is possible, therefore, to measure unique, individual enzymes during interactions with proteins. In fact, results like this are usual when imaging by electron microscopy semiconductors or other solid materials, because such samples are not damaged by interactions with accelerated electron beams in vacuum. Data obtained applying this workflow may allow to speed-up understanding of protein-protein interactions, leading to focus on effective drugs. A rationale of this behavior is proposed, stemming out of the discrete nature of interactions between high-energy electron beams and matter. Finally, this workflow offers the new possibility to store samples under liquid nitrogen after the first observation, for more imaging of the same area in more details; a characteristic useful for imaging large protein groups.
Here we document results obtained by electron microscopy tomography after increasing the resistance of flash-frozen viral proteins to accelerated electron beams, consequent to a few hours’ treatment in high vacuum at cryo-temperatures. Such temperature curing greatly reduces dose limitations, therefore allowing high-resolution tomography experiments also on flash frozen protein aggregates, and without the need of averaging multiple samples. In this way three-dimensional atomic scale images can be realized on one individual aggregate and in one experiment. This workflow has been tentatively introduced for flash-frozen rigid biopolymers, but only here this method is further generalized and applied to flash-frozen, deformable oligomers of proteins extracted from the HIV-1 virus. These results indicate that protein constructs can be imaged down to atomic coordinates by electron microscopy tomography without recurring to averaging among many samples. It will be possible, therefore, to measure unique, individual molecules also during the timed interaction with other proteins. In fact, results at this scale are usual with electron microscopy when imaging semiconductors or other solid materials, because such samples are not quickly damaged by interactions with accelerated electron beams in vacuum. In summary, data obtained applying this workflow could allow to speed-up significantly the understanding of protein- protein interactions, leading biological research to focus on effective drugs. A tentative rationale of this behavior is proposed, stemming out of the discreet nature of the interactions between high-energy electron beams and matter.
Finally, this workflow offers the possibility, new to cryo-electron microscopy of biologic samples, to store a sample indefinitely under liquid nitrogen after the first observation, for more imaging of the same area in more details. This characteristic may be useful for imaging complex tissues, or large groups of many proteins down to atomic resolution, like e.g. in brain tissue.
Keywords
Lymphatic filariasis, Medicinal plant, Traditional medicine
Introduction
The restricted stability of frozen biological samples to accelerated electron beams in vacuum has been the major limiting factor for high-resolution studies by Electron Microscopy (EM), since the introduction of fast-freezing immobilization techniques. While flash freezing allows to immobilize proteins in their instantaneous configuration, when imaging with accelerated electron beams samples degrade rapidly. Such situation has limited accurate determinations because EM signal follows Poissonian statistics, where the error is low enough only when the number of events (i.e. electrons scattered) is large enough. Later, averaging data from many thousands experiments have been introduced for by passing this problem, but the low signal-to-noise (S/N) of individual images used for calculating the average structures limit applications. Until today no viable solutions to increase the stability of such frozen samples under electron-beams has been proposed. Previously introduced only for rigid cellular biopolymers (Fera A, 2021), here results here will focus on electron microscopy of isolated, assembled aggregates of the R18L mutant of the CA matrix protein of the Human Immunodeficiency Virus (HIV-1). If confirmed, this behavior could allow to use this workflow for acquiring in one single experimental session structural data at sub-atomic resolution of flash-frozen viral proteins (Clementi E and Raimondi DL, 1963; Clementi E, et al., 1967).
Therefore atomic-scale structures of individual (not averaged) proteins, including when out-of equilibrium, would become available. Such advances may help the introduction of new drugs aiming at interfering with targeted mechanisms, like viral entry. The data presented here can be explained in the framework of the quantization of the energy of the atomic (electronic) states. Because of this quantization, it is widely accepted (Tavernier S, 2010) that the probability of interference of atomic electrons with accelerated incoming electrons (Pel) is inversely proportional to the difference between the kinetic energy of the incoming electrons (EkeV) and the ionization energy (E0) of the atomic electrons before interaction.
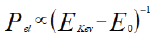
This dependence is the underlying motivation of the very high elemental sensitivity of modern chemical surface analysis techniques, like Auger spectroscopy. From equation, when the term between brackets grows, Pel decays monotonically.
Using this framework, results are shown here from an original workflow able to output flash frozen proteins resistant to sustained electron irradiation. The physical origin of this behavior remains a matter of debate. Here experimental findings are made known to the wide scientific community in order to accelerate its fruition and, hopefully, progress. A mere possible interpretation is attempted. Recently it has been demonstrated that a flash-frozen sample of rigid biopolymers (Fera A, 2021) (previously stained by exposing it to a transition metal salt), when inserted in high-vacuum conditions for a long enough time, while maintained at controlled temperatures (always well below freezing point), are able to sustain, unaltered, over 1.5 hours of illumination from a 120 keV electron beam. In fact, if confirmed, those data may shed light on the interactions of the individual subunits in solution, and on the mechanisms of formation of such biopolymers, in vitro. For the present work, that result sets the stage to test such stability on viral proteins (and oligomers) to sustained irradiation from high energy electron beams. Here we focus on a similar high-vacuum curing of assembled, flash-frozen protein constructs extracted from HIV-1, and maintained at all times at temperatures well below freezing inside a Transmission Electron Microscope (TEM) column, before illuminating with an accelerated electron beam. The proteins cured in this way are here demonstrated to become resistant to the rigors implicit to obtaining the high-S/N images necessary to reconstruct a high-resolution tomogram (i.e. able to show signal variations of the order of two voxels size). It is further shown that this behavior is independent of pre-staining the sample with heavy-metal cross-linkers. In these conditions, i.e. in the high-vacuum atmosphere inside a TEM, and virtually in absence of water molecules in the beam path, it has also been possible to image un-stained biological samples (albeit with a very sensitive camera) using a beam focused within ˜300 nanometers from zero defocus level. These results seem to indicate the possibility to use this method for studying directly (with low defocus) protein-protein interactions, like e.g. viral spikes interacting with cell receptors.
Materials and Methods
R18L mutants of the CA protein of HIV-1 have been kindly donated by Barbie Ganser-Pornillos and Owen Pornillos. They have been assembled in the buffer provided immediately before deposition on a cupper formvar or formvarcarbon grid, 200 or 300 mesh (EMS Sciences). The deposition has been realized suspending the grid (with the shiny side facing up) on a self-closing tweezer (EMS-Sciences). Using a standard pipette, a ˜6 μl drop containing the fresh assembled R18L CA protein has been deposited on a grid, on the bench, waiting about 1.5 minutes for the second step. Then, after wicking the excess solution, a ˜6 μl drop of 2% (w/v) Uranyl Acetate was deposited on the grid for about 45 seconds. After wicking the excess solution, another drop of solution containing 6 nm colloidal gold spheres is deposited on the sample for a minute, followed by mounting the sample vertical in the ‘guillotine’ routinely used in Dr. Dryden’s lab for quickly plunge-freezing samples by plunging in liquid ethane, after blotting carefully the excess solution from the vertical grid. Analogously, for the unstained sample, a ˜6 μl drop containing R18L assembled in exactly the same way has been deposited on a Formvar-C 200 mesh grid, for about 2 minutes, wicking the excess solution immediately before adding 6 μl of the same Asurion solution containing 6 nm colloidal gold. After about a minute, the grid with R18L has been mounted vertical in the aforementioned guillotine, wicking the solution in excess while vertical, and immediately plunge-freezing plunging in liquid ethane.
Once the samples were flash-frozen, they have been inserted in a Gatan Cryo holder and mounted inside the TF20 Tecnai, making sure that they were in contact with liquid nitrogen all the time. Once connected to the sample holder inserted in the TEM, the Gatan controller measured typically -172°C. Then a temperature ramping cycle has been started setting as final temperature -100°C at a speed of 3°C/min. When the set temperature was reached, or immediately before to prevent overshooting, the speed of heating has been set to 1°C/min and the final temperature has been set to -90°C or -92°C (Pornillos O, et al., 2011). Temperature has been maintained constant during EM imaging afterwards at varying angles, making sure to record as well a second image at 0° tilt at the end of the experiment, guaranteeing that the sample has not been altered by the sustained irradiation.
Tomograms have been acquired in two ways: (1) varying the angle by hand ± 60° acquiring high S/N images (like Figure 1) every 1°; (2) using the SerialEM software to vary the angle in the same range. This allowed to standardize some settings like average defocus, which was set at -300 nm in the unstained experiment reported (Figures 2 and 3). Each image used for calculating a tomogram has been acquired with at least 8000 e-/ nm2. Therefore, including also the images for centering, a complete tomography acquisition is realized with an electron dose of the order of 1.4106 e-/nm2. After imaging, samples have routinely been stored under liquid nitrogen. When necessary, the same area has been re-imaged the next day, sometimes directly at liquid nitrogen temperature if no significant ice was deposited on the sample during handling outside the electron microscope. All samples imaged are stored under liquid nitrogen in the facility of the medical school of UVA in Charlottesville, VA, where all experiments have been carried out at the time of writing this manuscript.

Figure 1: Successive images of the same assembled R18L-CA protein at different stages of the tomography acquisition. (Left) at the beginning; (right) after a full acquisition for a tomogram with 121 images.

Figure 2: Perpendicular virtual sections extracted from a tomogram calculated from imaging as described an individual positive stained assembled construct of the R18L-CA-HIV-1 protein. Left: XY plane; Center: XZ plane and Right: YZ plane. The yellow cross indicate the same point in the three planes for clarity. Contrast inverted. Image unbinned, voxel size: 0.1745 nm.
Results
Figure 1 shows two images of the same grid with deposited assembled mutant R18L of the CA protein of the HIV-1 capsid, kindly donated by Owen Pornillos and Barbie Ganser-Pornillos of the Medical School at the University of Virginia (UVA) in Charlottesville, VA. All images shown here have been obtained with a non-aberration-corrected TEM, operating at 200 keV at the modern TEM facility of the Medical School of the UVA in Charlottesville (VA). The data shown in Figures 1 and 2 have been obtained using a magnification leading directly a pixel size of 0.1745 nm. The two images in Figure 1 have been obtained, respectively, at the beginning of the tomography series (left), and after 121 images at varying angles (right). All images of stained samples reported (as Figure 1) here have been obtained manually focusing at increasing tilt angles.
From a closer analysis of the images, apart for differences in defocus, it is apparent that the two images show the same details. The area shown is a small part of the 4096 × 4096 field of view, in order to appreciate the signal invariance at high magnification. Note the 6 nm colloidal gold beads used as fiduciary markers. Since each image had to be focused manually, small differences of defocus were uncontrollable at the time of this experiment. More trials with automatic software are on the way but standardizing defocus level is not obvious with the precision required here. Our current interpretation is that this stability of the proteins irradiated is a consequence of the curing prior to illumination with the electron beam. Figure 2 shows (with inverted contrast) three perpendicular virtual sections (from left to right, respectively, the XY, the XZ and the YZ) calculated from the corresponding tomogram of the object indicated by a red arrow in Figure 1. Figure 3 shows analogously XZ, XY and YZ virtual sections extracted from another tomogram measured from an unstained assembled aggregate of the same R18L mutant of CA-HIV-1 (Pornillos BKG, et al., 2004; Pornillos BKG, et al., 2007), imaged and shown with magnification leading directly to a pixel size of 0.1112 nm. The pictures demonstrate that as well this sample, subjected to the same temperature curing preceding illumination, is not collapsed (although Figure 3 is shown binned 4X). For comparison, a tomogram of the same constructs of R18L-CA-HIV-1 negative stained and air-dried on grids, shows collapsed structures of ˜7 nm height (data not shown), as found before for negative stained enveloped virus (Pornillos O, 2011). Figures 2 and 3 show structures clearly not collapsed while corresponding renderings (Figures 4 and 5), realized fixing a unique threshold for the iso surface, reveals the typical hexagonal motif of this protein. Held together by dipole-dipole interactions, the hexagonal motifs in these samples are conserved especially in the shape of the central cavities of the constituting ‘donuts’. The common factor in all experiments seems to be the fact that free water molecules sublimate during the curing inside the TEM column before illuminating with accelerated electrons (Fera A, et al., 2012; Zingsheim HP, 1984). This is accomplished always well below freezing point, but at temperatures significantly higher than the liquid nitrogen (materials and methods). The absence of ice was indicated by the absence of opaque areas that progressively disappear when illuminated by the electron beam. When ice was still present, those areas were not chosen for further elaboration.

Figure 3: Perpendicular Virtual sections (XZ, XY and YZ planes respectively) extracted from a tomogram measured imaging an unstained individual aggregate of the R18L mutant of the CA-HIV-1 protein. Images acquired at pixel size 1.112Å but shown after binning by 4 using the scale function of Fiji-ImageJ. The yellow marker indicates the same point in the three projections. A fiduciary marker is seen in the XY projection. Contrast inverted. Scale bars: 10 nm.

Figure 4: 3D rendering of the aggregated R18L mutant of CA-HIV-1 protein, imaged unstained and shown in Figure 3; this sample has been imaged at -90°C. The binning 4X results in a smoother surface than the corresponding stained sample. Note the characteristic size and shape of the holes of the constituting units of this aggregate determined by the strong dipole-dipole interactions between monomers.

Figure 5: 3D rendering of the R18L mutant of the assembled CA- HIV-1 shown in figure 1 and 2. This sample has been measured after staining with UA followed by a quick washing step before flash-freezing; Tomogram acquired at -92°C. The known size and shape of the hexagonal motives is recognizable (please use circles as guides). The blue cube is meant as size reference: side=2 nm. It has been placed roughly in the same plane as the objects inside the red circle.
In order to complete the present illustration of results, Figures 4 and 5 show corresponding 3D renderings from the tomograms acquired with the samples shown above. These renderings have been obtained with the freeware software TOMVIZ (Cornell and Michigan University), using the ‘contour’ tool that renders iso-surfaces.
Discussion
At present, the physical origin of these surprising results is not clear, and some artifacts may not be completely ruled out at this stage. The main, current interpretation is that this discovery stems out and benefits from the instability of free water molecules in vacuum, where they spontaneously sublimate at these temperatures. In fact, water molecules not bound chemically to (polar) molecular groups are well-known to sublimate at the temperatures used during curing (Fera A, et al., 2012; Zingsheim HP, 1984) (and just a slower at liquid nitrogen temperatures) when in a high-vacuum environment, as the environment inside a TEM column. In these conditions, proteins stay solid while water sublimation can reach a rate of a few nm/minute (Fera A, et al., 2012). At the current level of understanding, such considerations may contribute as well to tentatively explain some behaviors commonly observed when imaging (unstained) flash-frozen but hydrated biological samples. In fact, unbound water molecules likely increase the rate of absorption of the parasitic radiation generated by the electron beam while passing through a biological sample. Indeed, considering the energy variation experienced by electrons deflected while penetrating a cryo-biological sample, the biggest majority of events (Tavernier S, 2010) involves very small energy exchanges, likely of the order of 1 eV or a fraction. Actually, 1 eV corresponds to 6000 cm-1, which is actually about double the center of the strongest (not unique) Raman absorption peak for water molecules (Wagner W and Riethmann T, 2011; Pastorczak M, et al., 2008). Water indeed absorbs strongly at Raman frequencies, which results in atomic vibrations. This strong Raman adsorption of water is not shared by R-OH groups, interpreted as a consequence of their different symmetry. This tentative theory may suggest a reason why ice degrades (melts) rather quickly after illumination with an electron beam. Instead, following this reasoning, isolated proteins would remain almost untouched by the electron beam, which suggests the two images compared in Figure 1.
For the sake of argument, if adopting this reasoning, the higher bound (i.e. the high-temperature limit where this attempted mechanism would lose validity) would be where the proteins are not rigid anymore, i.e. at their melting temperature. In other words, temperature has to be kept at all times well below the melting point of proteins, in order to guarantee the absence of obvious distortions which does not happen where ice melts to water. Anyhow, for all experiments shown here, temperature has been kept at all times below or around (minus)-90°C. Which value has been chosen studying the phase diagram of ice and the known data pertaining the speed of sublimation of ice, in vacuum, well below 0°C, a situation incurred at times in studies of interstellar space or planets without an atmosphere (Fera A, et al., 2012; Zingsheim HP, 1984). It is well-known that the interaction of accelerated electrons with an atom is not a continuous spectrum at all energies of the incoming electrons. It is instead a discrete sequence of absorption lines (e.g. Auger Spectroscopy) depending on the energy of the incoming electrons, which has to match the ionization potential of one atom in the material invested by the radiation. In other words, Pel has to always be taken into account (Tavernier S, 2010) since the atomic electron energy levels are quantized. Therefore, examining the energy levels of the inner core electrons of the heaviest element in these samples, Uranium has the 1s core energy level in the range of ˜105.600 keV (Jancso G, et al., 1970). Only electrons with energies within a few percent will be able to interact with the 1s electrons of Uranium, and the corresponding level in Uranyl cannot be at much higher energies since, when using 120 keV electrons, no interaction has been observed (Fera A, 2021). All other elements in these samples have electron levels of less energy, so further from the energy of the electrons in the beam. Other possible candidates are the energy levels of the nuclei, at least at 3 orders of magnitude higher. In summary, elementary physics models (Tavernier S, 2010) seem to predict that electrons accelerated at 200 keV would pass through this sample with only elastic scattering events. As may be indicated by the possibility of measuring the same image after 1.5.106 e–/nm2 (Figure 1) (Hobley DEJ, et al., 2013).
Conclusion
In summary, this invariance seems to be due to the fact that no energy levels in the sample are near 200 keV, i.e. the energy of the accelerated electrons from the electron microscope used. One other remarkable fact stemming out of these trial measurements is that unstained, unaveraged protein molecules can be observed in vacuum without high defocus, at least when virtually in absence of free water. The details shown in Figure 4 are of the order of 0.5 nm, as evinced from the central cavities of the hexagonal units at the center of every ‘donut’ in the renderings of Figure 4, whose data file is binned 4X. Without binning, the rendering of Figure 5 shows a structure with higher level of details but, notably, very similar central cavities of the motifs. As expected, since such interactions are strong dipole-dipole interactions, and they are stronger than the thermal energy at room temperatures. Further elaborations of similar samples, and possibly some averaging, may allow to better reproduce the underlying symmetry readily found by cryo-EM.
Acknowledgment
All the experiments here reported have been carried out using the microscopes and equipment/personnel of the Medical School in the University of Virginia Charlottesville, assisted by Dr. K. Dryden. The very kind, prompt response of the Ganser-Pornillos group (especially Barbie and Owen) has allowed this work to be completed in a rather short time. AF acknowledges the help of Mira Shapiro refining the grammar of this manuscript. For discussions at various stages of the preparation of this manuscripts AF acknowledges Dr. P. Ercius, of the Molecular Foundry at the Lawrence Berkeley National Laboratory in Berkeley, California. As well, Dr. A. Smirnov is acknowledged for critical reading of this manuscript and for many discussions dating the early stages of conceptualization of this work.
References
- Fera A. The endurance of flash-frozen biologics can be improved allowing high-resolution electron microscopy tomography on individual proteins. Microsc Microanal. 2021; 23(Suppl 1): 1114-1115.
- Clementi E, Raimondi DL. Atomic screening constants from SCF functions. J Chem Phys. 1963; 38: 2686.
- Clementi E, Raimondi DL, Reinhardt WP. Atomic screening constants from SCF functions. II. atoms with 37 to 86 electrons. J Chem Phys. 1967; 47: 1300.
- Tavernier S. Experimental techniques in nuclear and particle physics. Springer. 2010.
- Pornillos BKG, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. Assembly properties of the human immunodeficiency virus type 1 CA protein. J Virology. 2004; 78(5): 2454-2464.
[Crossref] [Google scholar] [Pubmed]
- Pornillos BKG, Cheng A, Yeager M. Structure of full-length HIV-1 CA: A model for the mature capsid lattice. Cell. 2007; 131(1): 70-79.
[Crossref] [Google scholar] [Pubmed]
- Pornillos O, Ganser-Pornillos BK, Yeager M. Atomic-level modelling of the HIV capsid. Nature. 2011; 469(7330): 424-427.
[Crossref] [Google scholar] [Pubmed]
- Fera A, Farrington JE, Zimmerberg J, Reese TS. A negative stain for electron microscopic tomography. Microsc Microanal. 2012; 18(2): 331-335.
[Crossref] [Google scholar] [Pubmed]
- Zingsheim HP. Sublimation rates of ice in a cryo-ultramicrotome. J Microsc. 1984; 133(3): 307-312.
[Crossref] [Google scholar] [Pubmed]
- Wagner W, Riethmann T. New equations for the sublimation pressure and melting pressure of H2O ice Ih. J Phys Chem Ref Data. 2011; 40(4): 043103.
- Pastorczak M, Kozanecki M, Ulanski J. Raman resonance effect in liquid water. J Phys Chem A. 2008; 112(43): 10705-10707.
[Crossref] [Google scholar] [Pubmed]
- Jancso G, Pupezin J, van Hook WA. The vapor pressure of ice between +10-2 and -1020. J Phy Chem A. 1970; 74(15): 2984.
- Hobley DEJ, Moore JM, Howard AD. How rough is the surface of Europa at lander scale? In Lunar and Planetary Science Conference. 2013; 1719: 2432.
Author Info
Andrea Fera*Citation: Fera A: High-Resolution Electron Microscopy Tomography of Interacting Flash-Frozen Proteins
Received: 15-Dec-2021 Accepted: 29-Dec-2021 Published: 05-Jan-2021, DOI: 10.31858/0975-8453.13.1.48-51
Copyright: This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
ARTICLE TOOLS
 Abstract
Abstract- Dental Development between Assisted Reproductive Therapy (Art) and Natural Conceived Children: A Comparative Pilot Study Norzaiti Mohd Kenali, Naimah Hasanah Mohd Fathil, Norbasyirah Bohari, Ahmad Faisal Ismail, Roszaman Ramli SRP. 2020; 11(1): 01-06 » doi: 10.5530/srp.2020.1.01
- Psychometric properties of the World Health Organization Quality of life instrument, short form: Validity in the Vietnamese healthcare context Trung Quang Vo*, Bao Tran Thuy Tran, Ngan Thuy Nguyen, Tram ThiHuyen Nguyen, Thuy Phan Chung Tran SRP. 2020; 11(1): 14-22 » doi: 10.5530/srp.2019.1.3
- A Review of Pharmacoeconomics: the key to “Healthcare for All” Hasamnis AA, Patil SS, Shaik Imam, Narendiran K SRP. 2019; 10(1): s40-s42 » doi: 10.5530/srp.2019.1s.21
- Deuterium Depleted Water as an Adjuvant in Treatment of Cancer Anton Syroeshkin, Olga Levitskaya, Elena Uspenskaya, Tatiana Pleteneva, Daria Romaykina, Daria Ermakova SRP. 2019; 10(1): 112-117 » doi: 10.5530/srp.2019.1.19
- Dental Development between Assisted Reproductive Therapy (Art) and Natural Conceived Children: A Comparative Pilot Study Norzaiti Mohd Kenali, Naimah Hasanah Mohd Fathil, Norbasyirah Bohari, Ahmad Faisal Ismail, Roszaman Ramli SRP. 2020; 11(1): 01-06 » doi: 10.5530/srp.2020.1.01
- Manilkara zapota (L.) Royen Fruit Peel: A Phytochemical and Pharmacological Review Karle Pravin P, Dhawale Shashikant C SRP. 2019; 10(1): 11-14 » doi: 0.5530/srp.2019.1.2
- Pharmacognostic and Phytopharmacological Overview on Bombax ceiba Pankaj Haribhau Chaudhary, Mukund Ganeshrao Tawar SRP. 2019; 10(1): 20-25 » doi: 10.5530/srp.2019.1.4
- A Review of Pharmacoeconomics: the key to “Healthcare for All” Hasamnis AA, Patil SS, Shaik Imam, Narendiran K SRP. 2019; 10(1): s40-s42 » doi: 10.5530/srp.2019.1s.21
- A Prospective Review on Phyto-Pharmacological Aspects of Andrographis paniculata Govindraj Akilandeswari, Arumugam Vijaya Anand, Palanisamy Sampathkumar, Puthamohan Vinayaga Moorthi, Basavaraju Preethi SRP. 2019; 10(1): 15-19 » doi: 10.5530/srp.2019.1.3