




Google Scholar citation report
Citations : 25586
Systematic Reviews in Pharmacy received 25586 citations as per google scholar report

Systematic Reviews in Pharmacy peer review process verified at publons

Indexed In
Review Article - (2022) Volume 13, Issue 5
Abstract
Presently, topical preparations have grabbed researchers' attention and proved to be the safest and most patient compliant drug delivery systems. Still, some gaps must be filled to get higher penetration and effectiveness of topical drug delivery systems. The present review focuses on one such approach, which has been proven as a promising tool to enhance the solubility of BCS class II drugs and improve drug permeation rate through the skin. The formulation contains the drug with higher energy and rapid dissolution in a solvent. The solution must have to be stabilized for complete absorption and bioavailability. Therefore, supersaturated solutions are prepared by adding precipitation inhibitors to prevent crystal growth. The solubilizing agents are used to incorporate higher drug amounts in a supersaturated state. This system provides the highest permeation rate of the drug as compared to other systems in topical preparations. This article will help the researchers effectively use the supersaturated drug delivery system for newer classes of drugs to incorporate into topical preparations.
Keywords
Supersaturation, Precipitation inhibitors, Topical drug delivery, Permeation enhancer
Introduction
For the last few decades, pharmaceutical research and industry have elucidated numerous innovations and practices in pharmaceutical formulations. Besides the traditional oral solids, several novel drug delivery systems such as topical formulations open the doors for more comfortable and patient-friendly preparations (Nastiti CMRR, et al., 2017). However, fast track commercialization of topical preparations requires thoughtful, open discussion of broader societal impacts.
Selection of the optimum formulation to get the desired action is the only challenge in novel formulation development. The topical route of administration of drugs is a safe, effective and patient compliant approach, which delivers drugs directly to the affected area on the skin (Chavan P, et al., 2016).
When taken orally, numerous drug candidates exhibit adverse effects like gastric mucosa irritation, hepatic first-pass metabolism, and degradation in an acidic environment (Derry S, et al., 2015; Massey T, et al., 2010). Therefore, topical delivery is the most emerging segment of pharmaceutical research, which exhibit many advantages, including patient compliance by avoiding above discussed side effects of the drug. Furthermore, topical formulations like gel, ointment, powders etc., and topical sprays give easy application, quick onset of action, and are delivered in predetermined meter dose containers (Chavan P, et al., 2016; Bakshi A, et al., 2008; Lu W, et al., 2013).
Literature Review
Human skin
The skin has been referred to as the most significant body organ: An average adult's skin has a surface area of about 2 m2, one-third of the total body (Spencer TS, 1988; Sévrain VS and Bonté F, 2007). The absorption of the drug occurs via the Stratum Corneum (SC), which is made up of dead, keratinized epidermal cells. The thickness of SC is ten μm, and it acts as a barrier to the permeation of drugs. The topical drug delivery system is an emerging field in the pharmaceutical sector, and these formulations face various challenges during the development and its smooth execution at the commercial level. Significant challenges of the formulator are poorly soluble drugs and poor penetration rate due to the SC layer of the skin.
The human skin structure (Figure 1) can be classified as follows (Ali S, et al., 2015).

Figure 1: Structure of skin
Taxonomical classification
• Microscale, which consists of cells and layers
• Mesoscale is consisting hair, freckles, wrinkles etc.
• Macroscale possesses body regions and body parts Histological classification
1. Epidermis
• Stratum basal (basal cell layer)
• Stratum spinosum (prickle cell layer)
• Stratum granulosum (granular cell layer)
• Stratum lucidum (clear layer)
• Stratum corneum (horny layer)
2. Dermis
• Papillary layer
• Reticular layer
3. Hypodermis
The drug penetration to the skin by mainly the three routes (Figure 2):

Figure 2: Routes of skin permeation (Parhi R, 2018)
• Intercellular route-The drug molecules diffuse from the gaps between two cells
• Transcellular route-The drug molecules pass through the cells by the saturation mechanism
• Transappendageal route-The drug molecule penetrates through sweat glands and hair follicles
The discovery of newer formulations frequently encounters in poorly water-soluble drugs. In most cases, these BCS class II drugs limit dissolution rate and affect drug absorption and bioavailability in oral formulations and lower diffusion in topical preparations. The problem demands novel and innovative techniques to obtain safe, efficacious and quality products. The following various methods modify the stratum corneum's barrier properties to enhance drug penetration through the skin.
• Chemical enhancement
• Physical enhancement
• Biochemical enhancement
• Bioconvertable prodrug
• Supersaturation enhancement
Supersaturated Drug Delivery System (SSDDS)
Solubility is the unique property of solute (solid, liquid and gaseous substances) to dissolve in solvent (solid, liquid and gaseous substances) to form a homogeneous solution (Brouwers J, et al., 2009). There are many factors like temperature, pressure; pH etc. affects the extent of solubility.
According to Biopharmaceutical Classification System (BCS), all drug candidates fall under four different classes (Moser K, et al., 2001).
• Class I=high solubility and high permeability
• Class II=low solubility and high permeability
• Class III=high solubility and low permeability
• Class IV=low solubility low permeability
Unfortunately, most drugs (more than 50%) available for pharmaceutical development belong to BCS Class II drugs. They are practically insoluble in water, which leads to inadequate and variable dissolution and bioavailability.
The marketability of poorly soluble drugs is the biggest question for pharmaceutical developers. Numerous methodologies and their practicability improve drug solubility and drug dissolution. Solubility improvement techniques involve physical modification like particle size reduction, crystal habit modification, co-crystallization, solid dispersion, cryogenic methods, and chemical modification like change in pH and surfactant and buffers (Schultz HB, et al., 2009; Press D, 2017).
The Supersaturated Drug Delivery System (SSDDS) is a promising approach to delivering high energy, rapidly dissolving drugs, which give adequate absorption and bioavailability. The strategy involves valuable and easy to prepare formulations, which can be stabilized by adding precipitation inhibitors. In addition, the SSDDS approach necessitates us to understand factors affecting the drug delivery system and drug's behaviour in the system.
For topical DDS, drug permeation is directly proportional to delta concentration across the skin and surface area covered by the dosage form (Parhi R, 2018; Maniyar MG and Kokare CR, 2018). A Supersaturated Drug Delivery System (SSDDS) is a novel and effective method, which can achieve high drug flux in topical DDS (Brouwers J, et al., 2009).
Supersaturated formulations are thermodynamically stable solutions, which encourage a higher concentration of dissolved solutes. In SSDDS, drugs exist in a solution state at a concentration above its saturation solubility (Rai V and Raghavan L, 2015). In this dosage form, maximum skin penetration rate without interfering barrier properties of stratum corneum layer and higher drug flux up to five to ten folds can be achieved due to the most increased thermodynamic activity of the drug (Brouwers J, et al., 2009; Schultz HB, et al., 2017; Guan J, et al., 2019) (Figure 3). Additionally, water is absorbed from the skin to the vehicle, increasing thermodynamic activity by acting as an anti-solvent in SSDDS.

Figure 3: Total versus free drug concentrations in suspension, solubilization, and supersaturation systems (Brewster ME, et al., 2008)
A supersaturated system is a more dissolved solute, ordinarily present in a saturated solution at a constant temperature. The spontaneous formation of crystals is considered a labile system (thermodynamically unstable), while in metastable state, solute remains in supersaturated solution without crystallization. It is maintained until the addition of foreign solid particles (e.g., Ultrasound). The above mention difference between labile and metastable state is known as Critical degree of saturation (Brouwers J, et al., 2009; Iervolino M, et al., 2000).
Higuchi first introduced the supersaturation concept in 1960. He stated that the rate-limiting step for absorption of drug residues in the outer layer of the Stratum Corneum (SC). According to partition coefficient, when a vehicle saturates with API applied on the skin surface, it leads to saturation of SC with the same API. Higher than this saturation level of API in vehicles leads to an increase in API amount in SC.
Further, it leads to enhancement of drug permeation in the same level of degree of saturation. The above concept is also based on concentration gradient. However, this is only feasible if the lipid in SC permits the drug to remain in a higher concentration, i.e., antinucleating agent.
Mechanism of supersaturation
As the thermodynamic activity of formation increases, the drug flux from the skin also increases due to supersaturation. This process occurs without affecting the skin's barrier and thus reduces the side effects or irritation (Nayak AK and Panigrahi PP, 2012; Mathews CDC and Sugano K, 2010).
The degree of saturation is expressed by supersaturation ratio (S)-
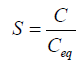
Where, C=drug concentration
Ceq=drug saturation concentration (Equilibrium solubility)
It is also expressed by relative supersaturation index (σ) or Degree of Supersaturation (DS)
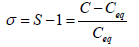
If, S<1 (σ<0)=Unsaturated
S=0 (σ=0)=Saturated
S>1 (σ>0)=Supersaturated
Supersaturated drug solution will return to the equilibrium state by drug
precipitation as it is thermodynamically unstable (Murthy SN and Shivakumar HN, 2010).
Guzman proposed the classical "SPRING" and "PARACHUTE" theory (Figure 4), which explains the development and preservation of metastable supersaturated states (24). Here, he explained SPRING means a high energy form of the drug in solution or solid form. SSDDS is expected to maintain the increased concentration of API for a more extended period at the application site to achieve more bioavailability. However, when supersaturation reaches, drug molecules tend to precipitate, which must be required to inhibit various Precipitation Inhibitors (PPIs). The mechanism of inhibition of precipitation is considered PARACHUTE. Solutions do not produce precipitations readily as they contain bound drugs within solvents, and their concentration is lower than the high equilibrium solubility (Figure 5a). On the other side, supersaturation exceeds concentration above its equilibrium solubility. Consequently, the state of supersaturation must remain constant even after preparation, as it is kinetically unstable preparation (Figure 5b). The supersaturated state transfers high-energy molecules to a lower energy state.

Figure 4: Spring and parachute approach

Figure 5a: Comparison of concentration profiles for solubilized, supersaturating systems and suspension

Figure 5b: Phase transition occurs in supersaturation, which is generated by dissolving an amorphous solid dispersion
Advantages of SSDDS
• A high permeation rate is achieved since the drug loading is usually higher in supersaturated solutions than other formulations (Parhi R, 2018; Rai V and Raghavan L, 2015).
• It has better physical formulation stability as it is monophasic (supersaturated solutions) than other biphasic (microemulsion) formulations.
• Supersaturated solutions can be prepared using indigenous material, and preparations are effortless, easy to scale up in industries.
Disadvantages of SSDDS
• As compared to other dosage forms like oral and parenteral, this system has low bioavailability.
• In SDDS, selecting the proper solvent system is challenging in the formulation because the thermodynamic effect depends on the chosen solvent system (Lu W, et al., 2013).
• It must be required to select proper API and other excipients with suitable drug response profiles for this type of formulation.
• In SDDS, formulation of drug crystallization is problematic, which can be overcome by the addition of antinucleating agents which inhibit nucleus formation.
Method for development of supersaturated solution
There are many methods to create supersaturation, like adding ions to initiate precipitation, solvent removal, pH and temperature change, dissolution of a metastable solid phase (Ng KW, 2018). However, co-solvency, pH change, solvent casting and heating-cooling cycle are popular methods for the same.
(i) pH change: This method is used for the weak base because weakly basic compounds exert higher solubility in the ionized form compared to unionize form. Supersaturation can be created by changing the pHs of the solution from lower than the pKa to higher. However, the pH change method disadvantages dilution with aqueous media (having pH at which compound is less soluble), forming precipitation (Ng KW, 2018).
(ii) Solvent casting method: This method is also known as the solvent evaporation method. In this method, solvent evaporation is followed by dissolution of the drug in PPI containing solvent like alcohol, acetone etc. However, the necessity of volatile solvents and precipitation during evaporation are significant downsides of the method.
(iii) Heating and cooling: When formulation was heated and cooled to room temperature, we obtain a solution having increase drug concentration above its solubility. The drug will be added to the solvent, suspended at 600 rpm, heated at 70°C for 10 min, and cooled at 23 ± 2°C. As the temperature reached below 25°C, 0.5 ml of sample is taken. The remaining solution is heated and cooled again. Perform the exact three times. First, the taken sample was centrifuged at 21,380 × g and 37°C 15 min. Next, take supernatant and again centrifuge and analyze in HPLC. Due to the high drug amount needed, these formulation and stability tests were conducted once (Koehl NJ, et al., 2020).
(iv) Evaporation of solvent: SDDS can prepare by evaporating solvent, which is effective for volatile solvents. By applying this formulation on the skin, the volatile solvent evaporates; thus, a decrease in solubility of residual solvent leads to the formulation of the supersaturation state of API. This system consists of two components-drug and solvent mixture, where solvent consists of volatile and non-volatile solvents (Edwards A, et al., 2017; Joshi P and Sangamwar AT, 2018). We have to select a drug, which is soluble in both types of solvent. Volatile solvent increases the concentration of the drug, while non-volatile solvent prevents precipitation from the solution. This method ensures sustained plasma drug concentration for a more extended period.
(v) Co-solvency: Mixtures of miscible solvents are called co-solvents. When the water-miscible solvent is used in this method, it changes the solubility of BCS class-II drugs, weakly electrolytes and non-polar molecules. The processes of increasing solubility of such drugs by the addition of water-miscible solvents are known as co-solvency. Co-solvents possess small hydrocarbon regions and hydrogen bond donor or/and acceptor groups. Co-solvency enhances solubility by different mechanisms like hydrophilic hydrogen-bonding groups, which improve water miscibility and hydrophobic hydrocarbon region, reducing the intermolecular attraction of water. Co-solvents disrupts water's self-association to increase solubility by decreasing its capacity to squeeze out hydrophobic or non-polar molecules (Nayak AK and Panigrahi PP, 2012; Ng KW, 2018).
Advantages of co-solvency compared to other techniques
• For formulations where increased solubility of the drug is necessary, this method enhances solubility up to several folds (Nayak AK and Panigrahi PP, 2012).
• The method is simple compared to the prodrug and salt formation method, where additional synthesis of new drug entities requires animal testing to confirm efficacy and safety.
• Methods having use of surface-active agents are toxic, but co-solvency is not.
• The rate and order of reaction can be changed by changing solvent properties.
• The chemical stability of drugs that undergo hydrolytic degradation is increased by decreasing water and using co-solvent.
• By facilitating an inadequate milieu for the transition state of the reactants, the stability of drugs is increasing.
• As compared to other methods, a high amount of drugs can be added and solubilize.
Theoretical considerations for characterizing dissolution and supersaturated behaviors under non-sink dissolution conditions
Supersaturation and passive absorption: According to several in-vitro experiments, the free drug concentration increases linearly as the solution becomes more supersaturated. Hence, the amorphous solubility of the drug-a maximum value of drug flux is obtained when the amorphous solubility of the drug is increased; however, the increase in a concentration above the maximum value does not increase the transport rate across the membrane. Thus, it is necessary to estimate the amorphous solubility of the drug (Ali S, et al., 2015; Rai V and Raghavan L, 2015).
Supersaturation and liquid-liquid phase separation: Liquid-Liquid Phase separation (LLPs) is obtained when amorphous solubility exceeds during dissolution. The phenomenon occurs when the kinetics of crystallization is slow during the dissolution process. LLPs occur only in non-sink condition, and another way to produce LLPs for the weakly basic compound is increased in solution pH upon dissolution of salts. This process depends on several factors like drug property, type of polymer, the particle size of dispersion, and media volume (Sun DD, et al., 2016). The compound with a high melting point is a thermodynamically stable form of crystallization as it acts as a precursor when drug-rich colloidal phase. Thus, it is expected to occur crystallization with depletion of supersaturation. Above the crystalline solubility, crystallization occurs. Upon increased concentration than the amorphous solubility, the possibility of LLPS or crystallization is higher. The colloidal drug-rich phase is persisted for several hours upon addition of crystallization inhibitors. LLPS will be obtained at a lower concentration as the ionic strength of the medium increases, but in the presence of surfactant, it will occur at a higher concentration. Thus, it is essential to note concentration where LLPS is obtained. LLPS also occurs in bio relevant media at higher concentrations than in buffer (Sun DD, et al., 2016).
Formulation aspects
The essential ingredients which are used in the formulation of SSDDS involve-
• Active Pharmaceutical Ingredients (API)
• Precipitation inhibitors
• Permeation Enhancer (PE)
• Solvent
Active pharmaceutical ingredient
Physicochemical properties:
• The molecular weight of chosen drug should be less than 1000 Daltons.
• Drugs have solubility in oil and water (log P should be 1-3)
• The drug should have an affinity for both lipophilic and hydrophilic phases.
• The melting point should be less than 200°C.
• The drug should have water solubility more significant than 1 mg/ml.
Biological properties:
• The drug should be potent with the use of small doses.
• The drug must have a short half-life.
• It should not be allergic or irritating.
• Drugs, which are degraded in GIT, can be used for this formulation.
•c The drug has to administer for an extended period.
Precipitation Inhibitors (PPIs)
Supersaturated formulations experience nucleation followed by precipitation. During the manufacturing of SSDDS, nucleations grow like a reciprocal of dissolution and coagulate drug molecules together in precipitation.
In the initial stage of crystallization, different cellulosic polymers (Hydroxypropyl methylcellulose, Hydroxypropyl cellulose, methylcellulose), rheological polymers (Polyvinyl pyrrolidone) and surfactants (Tweens, Chromophore etc.) act as PPIs and inhibit nucleation and crystal growth (Bannow J, et al., 2020; Ilie A, et al., 2020; Enin HAA and Abdel-bar HM, 2016).
There are three mechanisms involves in PPIs. Firstly, by decreasing surface tension, when surface tension decreases, it transfers the crystallization process to surface nucleation from diffusion control. Secondly, by increasing solubility, supersaturation decreases and subsequently, nucleation also reduce. Lastly, blocking crystal growth by crystal surface interfaces adsorbance may resist the availability of solutes to the crystal terrace. Adsorbance and disruption of growth steps may secure the availability of adsorbed particles to the crystal terrace, and the rough surface becomes flat, so there is no adsorbance into surface imperfections (Warren DB, et al., 2010).
Theory of drug precipitation
The difference in chemical potential (µ) between supersaturated and saturated solutions has indicated the driving force required for drug precipitation.
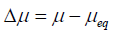
Where Δμ=difference of chemical potential
μ=potential energy of supersaturated solution
μeq=potential energy of saturated solution
From the definition of chemical potential, it follows that-
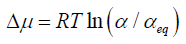
Where R = gas constant
T = temperature
α and αeq=the activity of solute in a supersaturated solution and saturated solution, respectively (Simonelli AP, et al., 1970).
If there is no difference in activity coefficient of solute of saturated state and supersaturated state, then the equation becomes zero
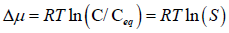
Where C=Concentration in the supersaturated solution
Ceq=It is the equilibrium solubility of the drug
S=Supersaturation ratio (Kashchiev D and Rosmalen GM, 2003)
Steps for drug precipitation
There are two steps consists of drug precipitation mechanism: nucleation and crystal growth. Nucleation in supersaturated solution means the dissolved amount of molecules come closer to each other and make the cluster of molecules. While this nucleation increases, they form crystals, which are called crystal growth or macroscopic crystal.
The precipitation that occurs from supersaturated solution is a thermodynamic process. The increase in Gibbs free energy cause precipitation because the nucleation step requires energy to aggregate molecule and form molecule cluster. The increased Gibbs free energy increases the interfacial tension and interfacial energy between solvent and molecule. If this energy is too high, no molecule aggregation or crystal will form; and the supersaturated solution achieves (Brouwers J, et al., 2009) (Figure 6).
Metastable zone
The amount of supersaturated concentration that can be existing, without the formation of precipitation is called a metastable zone. However, in the case of using precipitation inhibitors in a supersaturated solution, it will increase the metastable zone (Brouwers J, et al., 2009). Nucleation rate is the net production of critical cluster per unit time and unit of bulk volume.
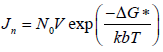
Where Jn=Nucleation rate
N0=The number of molecules in a unit volume
v=The frequency of molecular transport at the nucleus-liquid interface
kb=The Boltzmann's constant and
ΔG*=The Gibbs free energy change for the formation of critical clusters.
Mechanism of precipitation inhibitors
• By increasing solubility, reduction in the degree of supersaturation leads to decease precipitation
• By increasing viscosity of the formulation, reduction in mobility of molecules prevent nucleation
• By increasing particle-liquid interfacial energy, reduce nucleation
• Changing the adsorption layer at the crystal medium interface or changing in crystal habit interferes with nucleation and crystal growth.
• Increased level of solvent at crystal liquid interphase affects the integration of molecule into a crystal. Consequently, it prevents precipitation.
Factors affecting precipitation inhibitors
Majorly, chemical and physical interaction between drug and polymers reinforce precipitation inhibition, and these interactions are influenced by the following factors (Morrison J, 2021)-
Temperature: The weakening of intermolecular interactions between different molecules at high temperature increase solubility.
Molecular Weight (MW): Drug-polymer interaction becomes more potent with higher MW polymer, increasing viscosity and free functional groups on polymer chains.
Viscosity: Rate of drug diffusion via inhibition of crystallization in nucleation initiation and growth stage is decrease by increasing viscosity of the solution.
Dielectric constant: The dielectric constant is directly proportional to the interaction of drug and polymer and inversely proportional to solubility. So by decreasing the drug-polymer interaction, dielectric constant and subsequently increase solubility.
H-bonding: Increase the number of free hydrogen bonding sites may enhance the interaction of drug and polymer.
Examples of precipitation inhibitors
Different polymers are used as precipitation inhibitors like cellulose derivative (methylcellulose, Hydroxypropyl cellulose, and Hydroxypropyl methylcellulose), vinyl polymer (e.g., PVA, PVP, PVPVA) and ethylene polymer (e.g., PEG). The most widely used precipitation inhibitors are given in below Table 1 (Bakshi A, et al., 2008; Edwards A, et al., 2017; Joshi P and Sangamwar AT, 2018; Ilie A, et al., 2020; Warren DB, et al., 2010; Edwards A, et al., 2017; Lind M, et al., 2016; Ghosh I and Kohn BM, 2012).
| Name of precipitation inhibitors | Mechanism | Example of drug | Function |
|---|---|---|---|
| HPMC (0.5-5%) | Hydrogen bonding | Hydrocortisone acetate | To prevent nucleation and crystal growth |
| PVC (0.01% w/w) | Chemical bonding | Bicalutamide | Nucleation rate is not affected but interferes with a crystal growth rate |
| Surfactant (2.50%) TPGS
|
Solubility | Itraconazole | Stabilization time is 40 min (in TPGS), 5 min (in case of tween 20 and cremophor RH40) |
| Cyclodextrin -2.50%
|
Solubility | Itraconazole | Stabilization time at least 4 hours |
Note: TPGS is α-tocopherol methoxy polyethene glycol succinate, HP βCD is hydroxypropyl beta-cyclodextrin, and SBE βCD is sulfobutylether beta-cyclodextrin
Table 1: Examples of precipitation inhibitors
Permeation enhancers
Penetration enhancers also called absorption promoters and sorption accelerants are the substances used to increase the permeation of skin mucosa and the absorption of penetrant through the skin (Figure 7).
Theory of permeation enhancer
Diffusion is a passive kinetics process that follows a concentration gradient mechanism for moving molecules from higher concentration to lower concentration regions. Fick's first law can describe the steady-state diffusion (Ng KW, 2018). This equation describes the rate of transfer or flux or J depending on the membrane area, partition coefficient, and concentration gradient (Lane ME, 2013; Blaabjerg LI, et al., 2018). In this equation, negative sign indicted to decrease the concentration in the donor compartment.
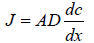
Where J=rate of mass transfer or rate of flux
A=area of membrane covered by applied formulation
D=diffusion coefficient
dc/dx=concentration gradient
Fick's second law of diffusion can be derived from the first law to describe membrane transport under non-steady-state conditions. This equation indicates a change in concentration with time leads to a change in concentration gradient.
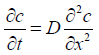
This equation can indicate a change in concentration profile with time after application of the given formulation. At the same time, the formulation is applied to the skin's surface at a maximum fixed concentration in the donor compartment and maintaining sink conditions in the receptor compartment. Therefore, this equation can be written as the following at a steady state.
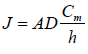
Where, cm=concentration of compound at donor membrane interphase h=effective diffusional pathlength of membrane
The obtained equation can be converted through the replacement of cm by K and Cv. Here, K is the ratio of the concentration of drug in the permanent membrane and donor compartment. This equation is descriptive of steadystate flux across the membrane. Therefore, with the help of Fick's law, the transport of molecules in the membrane can be understood.
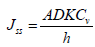
Where K=Vehicle membrane partition coefficient
Cv=Concentration of compound in donor compartment
The above equation indicates that the increased transfer rate depends on the change in D, K and cv. Therefore, permeation enhancers must change the solubility or partition drug behaviour into the SC.
Ideal characteristic of penetration enhancer
• Pharmacologically inert
• Non-toxic
• Non-irritant
• Fast-acting
• Non-allergic
• Predictable and reproducible duration of action
• Compatible with drug molecules and other excipients
• Cosmetically accepted
Merits of penetration enhancers
• Increase the therapeutic efficacy of the drug by increasing penetration of drug molecule
• Useful to penetrate un-absorbable drugs by facilitating their absorption
• Improve the absorption efficiency of topical formulation
• Do not affect zero-order skin permeation profile
• The terpenes are effectively used as penetration enhancers in cytotoxic drugs
Classification of penetration enhancers (Moser K, et al., 2001; Ng KW, 2018; Lane ME, 2013; Blaabjerg LI, et al., 2018)
The various types of penetration enhancers (Table 2) include chemical enhancers, physical enhancers, natural enhancers, drug vehicle, biochemical approaches and miscellaneous enhancers. The most commonly used chemical penetration enhancers in the pharmaceutical industry are shown in Table 3 with examples.
| Type of penetration enhancer | Mechanism of penetration enhancer | Example of penetration enhancer |
|---|---|---|
| Chemical enhancer |
|
|
| Drug vehicle-based penetration enhancer | Interaction with stratum corneum and development of SAR for enhancing with optimal characteristic and minimal toxicity |
|
| Natural penetration enhancers | Partition coefficient, diffusion coefficient, lipid extraction, drug solubility, macroscopic barrier perturbation, molecule orientation of terpenes molecule with the lipid bilayer |
|
| Physical enhancer | By electrical current, ions exchange, magnetic energy, ultrasound wave and thermal energy |
|
Table 2: Classification of penetration enhancers with mechanism and example
| Type of chemical penetration enhancers | Examples |
|---|---|
| Alcohol |
|
| Amides | Cyclic amides (Azone) |
| Esters |
|
| Ether alcohol | Transcutol® |
| Fatty acids | Lauric acid, linoleic acid, linolenic acid, myristic acid, oleic acid, palmitic acid, stearic acid and isostearic acid |
| Glycol | Dipropylene glycol, propylene glycol, 1,2-butylene glycol, 1,3-butylene glycol |
| Pyrrolidone’s | N-methyl-2-pyrrolidone, 2-pyrrolidone |
| Sulphoxides | Decylmethyl sulfoxides, Dimethyl sulfoxide |
| Surfactants |
|
| Terpenes |
|
Table 3: Chemical classification of penetration enhancers with example
Solvents
The solvents play a pivotal role in film formation by solubilizing drugs. In addition, they have an important impact on drug permeation. Commonly used solvents for topical and transdermal use are listed in Tables 4 and 5.
| Name of drug | Patch trade name | Penetration enhancers |
|---|---|---|
| Buprenorphine | BuTrans | Oleyl oleate |
| Estradiol | Alora | Sorbitan monoleate |
| Estradiol | Divigel | Ethanol and propylene glycol |
| Estradiol | Elestrin | Ethanol, ether, diethylene glycol monoethyl |
| Nitroglycerine | Minitran | Ethyl oleate, glyceryl monolaurate |
| Testosterone | Androgel | Isopropyl myristate |
| Oxybutynin | Anturol | Ethanol, ether, propylene glycol diethylene glycol monoethyl |
Table 4: Example of various penetration enhancers used in commercial topical products (Ng KW, 2018)
| Chemical class of solvent | Examples |
|---|---|
| Glycols | Propylene glycol, polyethylene glycol |
| Alcohol | Ethanol, butanol, isopropanol, benzyl alcohol, lanolin alcohol, fatty alcohol |
| Other solvents | Ethyl acetate, oleic acid, isopropyl myristate |
Table 5: Different types of solvent used in transdermal delivery of the system
Evaluation parameters of supersaturated drug delivery systems
Determination of crystal growth: The easiest and widely used method for the determination of crystal growth is using a microscope. The thin film of the formulation spreads on the slide, and the presence or absence of crystals can be observed (Press D, 2017; Brewster ME, et al., 2008). Different methods like X-Ray diffraction, DSC, microcalorimetry, isothermal heat conduction are also famous for crystal growth determination (Ranade S, et al., 2017; Soltanpour S and Shekarriz A, 2015).
Supersaturation index: Supersaturation index or degree of supersaturation is the ratio of initial drug concentration to the equilibrium drug concentration against the polymeric solution containing anti-precipitants (Blaabjerg LI, et al., 2018; Chaudhari SP and Dave RH, 2016; Vandecruys R, et al., 2007). The titration of the drug-containing solution against anti-precipitant containing solution gives precipitation at a specific time point.
To assess the physical stability of the formulation Δ% is calculated as per the given equation-Δ%=(Drug concentration at 5 minutes-drug concentration at 120 minutes)/Drug concentration at 5 minutes × 100
Discussion and Conclusion
Pharmaceutical research has expanded to the development of novel drug delivery systems offering therapeutic effectiveness and technological advancement. The idea of delivering drugs through the skin avoiding the gastrointestinal tract is quite old; however, improvement of powerful novel and innovative dosage forms always remains the thrust area of the researchers. Supersaturation is the novel approach to enhance the solubility of poorly water-soluble drugs. Topical formulations enhance patient compliance by overcoming shortfalls of oral formulation for the treatment of various diseases. The topical supersaturated formulations are designed to improve the absorption and bioavailability of poorly water-soluble drugs by using precipitation inhibitors, solubilization techniques, and permeation enhancers. The review article is helpful to attain and maintain the supersaturation state in a supersaturated solution. Developing a stable supersaturated product is the biggest challenge; however, commercialization of the same becomes useful. Therefore, extensive research and development efforts and innovative designs are required to achieve potential technology advantages.
References
- Nastiti CMRR, Ponto T, Abd E, Grice JE, Benson HAE, Roberts MS. Topical Nano and microemulsions for skin delivery. Pharmaceutics. 2017; 9(4): 1-25.
[Crossref] [Google scholar] [PubMed]
- Murthy SN, Shivakumar HN. Topical and transdermal drug delivery. Elsevier. 2010. 1-36.
- Chavan P, Bajaj A, Parab A. Topical sprays : Novel drug delivery system. Int J Pharm Chem. 2016; 2(2): 102-111.
- Derry S, Ra M, Gaskell H, Mcintyre M, Pj W. Topical NSAIDs for acute musculoskeletal pain in adults (Review). Cochrane Database Syst Rev. 2015; (6): CD007402.
[Crossref] [Google scholar] [PubMed]
- Massey T, Derry S, Moore RA, Mcquay HJ. Topical NSAIDs for acute pain in adults. Cochrane Database Syst Rev. 2010; 6: CD007402.
[Crossref] [Google scholar] [PubMed]
- Bakshi A, Bajaj A, Malhotra G, Madan M, Amrutiya N. A novel metered dose transdermal spray formulation for oxybutynin. Indian J Pharm Sci. 2008; 70(6): 733-739.
[Crossref] [Google scholar] [PubMed]
- Lu W, Luo H, Wu Y, Zhu Z, Wang H. Preparation and characterization of a metered dose transdermal spray for testosterone. Acta Pharm Sin B. 2013; 3(6): 392-9.
- Spencer TS. Dry skin and skin moisturizers. Clin Dermatol. 1988; 6(3): 24-28.
[Crossref] [Google scholar] [PubMed]
- Sévrain VS, Bonté F. Skin hydration: A review on its molecular mechanisms. J Cosmet Dermatol. 2007; 6(2): 75-82.
[Crossref] [Google scholar] [PubMed]
- Ali S, Shabbir M, Shahid N. The structure of skin and transdermal drug delivery system-A review. Res J Pharm Technol. 2015; 8(2): 103-109.
- Parhi R. Transdermal evaporation drug delivery system : Concept to commercial products. 2018; 8(4): 535-550.
[Crossref] [Google scholar] [PubMed]
- Brouwers J, Brewster ME, Augustijns P. Supersaturating drug delivery systems: The answer to solubility-limited oral bioavailability? 2009; 98(8): 2549-2572.
[Crossref] [Google scholar] [PubMed]
- Moser K, Kriwet K, Kalia YN, Guy RH. Enhanced skin permeation of a lipophilic drug using supersaturated formulations. J Control Release. 2001; 73(2-3): 245-253.
[Crossref] [Google scholar] [PubMed]
- Schultz HB, Kovalainen M, Peressin KF, Prestidge CA, Thomas N. Supersaturated silica-lipid hybrid oral drug delivery systems: Balancing drug loading and in vivo performances. 2019; 742-750.
[Crossref] [Google scholar] [PubMed]
- Press D. Supersaturable solid self-microemulsifying drug delivery system: Precipitation inhibition and bioavailability enhancement. 2017; 8801-8811.
[Crossref] [Google scholar] [PubMed]
- Maniyar MG, Kokare CR. Formulation and evaluation of spray dried liposomes of lopinavir for topical application. J Pharm Investig. 2018; 20(5): 724-736.
[Crossref] [Google scholar] [PubMed].
- Rai V, Raghavan L. Transdermal drug delivery systems using supersaturation. 2015; 151-161.
- Schultz HB, Thomas N, Rao S, Prestidge CA. Supersaturated silica-lipid hybrids (Super-SLH): An improved solid-state lipid-based oral drug delivery system with enhanced drug loading. Eur J Pharm Biopharm. 2017;
- Guan J, Liu Q, Liu J, Cui Z, Zhang X, Mao S. Elucidation of alginate-drug miscibility on its crystal growth inhibition effect in supersaturated drug delivery system. Carbohydr Polym. 2019; 115601.
- Brewster ME, Vandecruys R, Verreck G, Peeters J. Supersaturating drug delivery systems: Effect of hydrophilic cyclodextrins and other excipients on the formation and stabilization of supersaturated drug solutions. Pharmazie. 2008; 63(3): 217-220.
[Crossref] [Google scholar] [PubMed]
- Iervolino M, Raghavan SL, Hadgraft J. Membrane penetration enhancement of ibuprofen using supersaturation. 2000; 198(2): 229-238.
[Crossref] [Google scholar] [PubMed]
- Nayak AK, Panigrahi PP. Solubility enhancement of etoricoxib by cosolvency approach. ISRN Phys Chem. 2012: 1-5.
- Mathews CDC, Sugano K. Supersaturable formulations. Drug Deliv Syst. 2010; 25(4): 371-374.
- Ng KW. Penetration enhancement of topical formulations. Pharmaceutics. 2018; 10(2): 10-12.
[Crossref] [Google scholar] [PubMed]
- Koehl NJ, Henze LJ, Kuentz M, Holm R, Griffin BT. Supersaturated lipid-based formulations to enhance the oral bioavailability of venetoclax. Pharmaceutics. 2020; 12(6): 1-20.
[Crossref] [Google scholar] [PubMed]
- Edwards A, Qi S, Liu F, Brown MB, McAuley WJ. Rationalising polymer selection for supersaturated film forming systems produced by an aerosol spray for the transdermal delivery of methylphenidate. Eur J Pharm Biopharm. 2017; 114: 164-174.
[Crossref] [Google scholar] [PubMed]
- Joshi P, Sangamwar AT. Stabilizing supersaturated drug-delivery system through mechanism of nucleation and crystal growth inhibition of drugs. 2018; 9(1): 873-885.
[Crossref] [Google scholar] [PubMed]
- Sun DD, Wen H, Taylor LS. Non-sink dissolution conditions for predicting product quality and in vivo performance of supersaturating drug delivery systems. J Pharm Sci. 2016; 105(9): 2477-2488.
[Crossref] [Google scholar] [PubMed]
- Bannow J, Yorulmaz Y, Löbmann K, Müllertz A, Rades T. Improving the drug load and in vitro performance of supersaturated self-nanoemulsifying drug delivery systems (Super-SNEDDS) using polymeric precipitation inhibitors. Int J Pharm. 2020; 575: 118960.
[Crossref] [Google scholar] [PubMed]
- Ilie A, Griffin BT, Kolakovic R, Vertzoni M, Kuentz M, Holm R. Supersaturated lipid-based drug delivery systems-exploring impact of lipid composition type and drug properties on supersaturability and physical stability. Drug Dev Ind Pharm. 2020; 46(3): 356-364.
[Crossref] [Google scholar] [PubMed]
- Enin HAA, Abdel-bar HM. Solid super saturated self-nanoemulsifying drug delivery system (sat-SNEDDS) as a promising alternative to conventional SNEDDS for improvement rosuvastatin calcium oral bioavailability. 2016; 13(11): 1513-1521.
[Crossref] [Google scholar] [PubMed]
- Warren DB, Benameur H, Porter CJH, Pouton CW. Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: A mechanistic basis for utility. J Drug Target. 2010; 18(10): 704-731.
[Crossref] [Google scholar] [PubMed]
- Simonelli AP, Mehta SC, Higuchi WI. Inhibition of sulfathiazole crystal growth by polyvinylpyrrolidone. J Pharm Sci. 1970; 59(5): 633-638.
[Crossref] [Google scholar] [PubMed]
- Kashchiev D, Rosmalen GM. Review: Nucleation in solutions revisited. Cryst Res Technol. 2003; 38(7-8): 555-574.
- Morrison J. A tale of two drug delivery strategies: Simple solubilization or sophisticated supersaturation? Am Pharm Rev. 2016.
- Lind M, Nielsen KT, Schefe LH, Nørremark K, Eriksson AH, Norsgaard H, et al. Supersaturation of calcipotriene and betamethasone dipropionate in a novel aerosol foam formulation for topical treatment of psoriasis provides enhanced bioavailability of the active ingredients. Dermatol Ther (Heidelb). 2016; 6(3): 413-425.
[Crossref] [Google scholar] [PubMed]
- Ghosh I, Kohn BM. A comparative study of vitamin E TPGS/HPMC supersaturated system and other solubilizer/polymer combinations to enhance the permeability of a poorly soluble drug through the skin. Drug Dev Ind Pharm. 2012; 38(11): 1408-1416.
[Crossref] [Google scholar] [PubMed]
- Lane ME. Skin penetration enhancers. Int J Pharm. 2013; 447(1-2): 12-21.
[Crossref] [Google scholar] [PubMed]
- Blaabjerg LI, Grohganz H, Lindenberg E, Löbmann K, Müllertz A, Rades T. The influence of polymers on the supersaturation potential of poor and good glass formers. Pharmaceutics. 2018; 10(4): 1-14.
[Crossref] [Google scholar] [PubMed]
- Ranade S, Bajaj A, Londhe V, Babul N, Kao D. Fabrication of topical metered dose film forming sprays for pain management. Eur J Pharm Sci. 2017; 100: 132-141.
[Crossref] [Google scholar] [PubMed]
- Soltanpour S, Shekarriz A. Naproxen solubility in binary and ternary solvents of polyethylene glycols 200, 400 or 600 with ethanol and water at 298. 2 K-experimental data report and modelling. Phys Chem Liquids. 2015: 53(6): 37-41.
- Chaudhari SP, Dave RH. Evaluating the effects of different molecular weights of polymers in stabilizing supersaturated drug solutions and formulations using various methodologies of the model drug: Fenofibrate. J Pharm Sci Pharmacol. 2016; 2(3): 259-276.
- Vandecruys R, Peeters J, Verreck G, Brewster ME. Use of a screening method to determine excipients which optimize the extent and stability of supersaturated drug solutions and application of this system to solid formulation design. Int J Pharm. 2007; 342(1-2): 168-175.
[Crossref] [Google scholar] [PubMed]
Author Info
Dipika Chavda1*, Sagar Chavda1, Jaina Patel1, Pratik Chauhan1 and Tejal Gandhi22Department of Pharmacology, Anand Pharmacy College, Gujarat, India
Citation: Chavda D: Supersaturated Drug Delivery System: A Novel Technique to Overcome Lacunas of Topical Formulation
Received: 01-Apr-2022 Accepted: 29-Apr-2022 Published: 06-May-2022, DOI: 10.31858/0975-8453.13.5.324-333
Copyright: This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
ARTICLE TOOLS
 Abstract
Abstract- Dental Development between Assisted Reproductive Therapy (Art) and Natural Conceived Children: A Comparative Pilot Study Norzaiti Mohd Kenali, Naimah Hasanah Mohd Fathil, Norbasyirah Bohari, Ahmad Faisal Ismail, Roszaman Ramli SRP. 2020; 11(1): 01-06 » doi: 10.5530/srp.2020.1.01
- Psychometric properties of the World Health Organization Quality of life instrument, short form: Validity in the Vietnamese healthcare context Trung Quang Vo*, Bao Tran Thuy Tran, Ngan Thuy Nguyen, Tram ThiHuyen Nguyen, Thuy Phan Chung Tran SRP. 2020; 11(1): 14-22 » doi: 10.5530/srp.2019.1.3
- A Review of Pharmacoeconomics: the key to “Healthcare for All” Hasamnis AA, Patil SS, Shaik Imam, Narendiran K SRP. 2019; 10(1): s40-s42 » doi: 10.5530/srp.2019.1s.21
- Deuterium Depleted Water as an Adjuvant in Treatment of Cancer Anton Syroeshkin, Olga Levitskaya, Elena Uspenskaya, Tatiana Pleteneva, Daria Romaykina, Daria Ermakova SRP. 2019; 10(1): 112-117 » doi: 10.5530/srp.2019.1.19
- Dental Development between Assisted Reproductive Therapy (Art) and Natural Conceived Children: A Comparative Pilot Study Norzaiti Mohd Kenali, Naimah Hasanah Mohd Fathil, Norbasyirah Bohari, Ahmad Faisal Ismail, Roszaman Ramli SRP. 2020; 11(1): 01-06 » doi: 10.5530/srp.2020.1.01
- Manilkara zapota (L.) Royen Fruit Peel: A Phytochemical and Pharmacological Review Karle Pravin P, Dhawale Shashikant C SRP. 2019; 10(1): 11-14 » doi: 0.5530/srp.2019.1.2
- Pharmacognostic and Phytopharmacological Overview on Bombax ceiba Pankaj Haribhau Chaudhary, Mukund Ganeshrao Tawar SRP. 2019; 10(1): 20-25 » doi: 10.5530/srp.2019.1.4
- A Review of Pharmacoeconomics: the key to “Healthcare for All” Hasamnis AA, Patil SS, Shaik Imam, Narendiran K SRP. 2019; 10(1): s40-s42 » doi: 10.5530/srp.2019.1s.21
- A Prospective Review on Phyto-Pharmacological Aspects of Andrographis paniculata Govindraj Akilandeswari, Arumugam Vijaya Anand, Palanisamy Sampathkumar, Puthamohan Vinayaga Moorthi, Basavaraju Preethi SRP. 2019; 10(1): 15-19 » doi: 10.5530/srp.2019.1.3